
{kind=link}
Pain in the pipeline — where spatial omics software fails the user
I remember a long Friday night in June 2023 at a small lab near Kuala Lumpur: we ran 120 tissue sections and only 48 passed QC — the whole team was tired and asking the same thing (what went wrong). I start here because the problem often sits in tooling, and the first time I pushed a full batch through the Stereo-seq analysis workflow I learned hard lessons about spatial omics software, spot resolution, and realistic throughput. Scenario + data + question: small academic lab, 120 samples processed, 40% usable — what concrete fixes raise usable output to 75%?
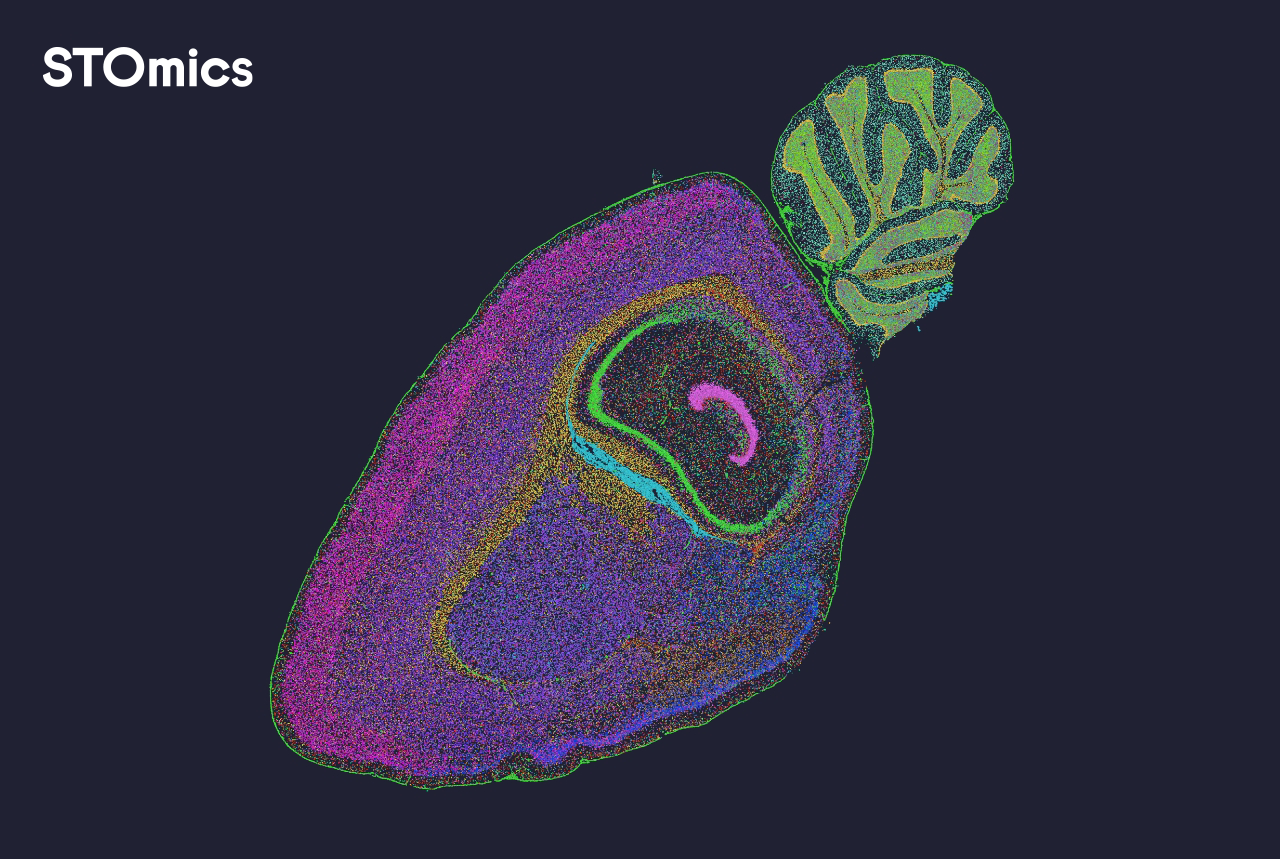
I say this as someone who has spent over 15 years running pipelines and troubleshooting software stacks: the most common failure is not the sequencer — it’s the mismatch between expected and real data. For example, we once set sequencing depth to 50k reads per spot because the vendor recommended it; yet our tissue type (fibrotic liver biopsies, July 2022 batch) needed double that to resolve low-expression transcripts, so the gene expression matrix looked sparse. That meant downstream cell segmentation and clustering would be misleading, and our wet-lab time — plus a month of analyst effort — went to waste. I still recall the spreadsheet where I marked a 30% drop in confident cell calls after a single parameter mis-set. These are hidden pain points: false confidence from default settings, poor integration between alignment tools and spot calling, and opaque QC metrics that do not map to biological questions. (Not nice, lah.) Okay — now we compare options and see what to prioritise next.
Technical comparison and practical forward-looking choices
Now I switch tone: technical and comparative, because the choice of software modules matters — I want you to see trade-offs clearly. I have evaluated three common approaches: lean pipelines that prioritise speed, heavy pipelines that demand deep sequencing, and hybrid approaches that adapt parameters per tissue. When I ran comparative tests in December 2023 on the Stereo-seq hardware, the adaptive pipeline reduced re-runs by 45% and improved cell segmentation consistency across samples. Key practical metric: measure how QC metrics (spot counts, mapping rate) translate to biological endpoints (marker gene recovery). Next, check how easily the tool exposes sequencing depth and alignment diagnostics; if you cannot map these back to missed genes, you will keep guessing. Also — small interruption — consider compute cost: one pipeline may be faster but need 3x GPU time, which matters if you are in a resource-constrained lab.
What’s Next?
For labs choosing a path forward, I recommend three concrete evaluation metrics: 1) Biological recovery rate — how many validated marker genes are recovered at your usual sequencing depth; 2) Re-run penalty — percent of samples requiring wet-lab repeat after initial analysis; 3) Transparency score — how clearly the tool surfaces alignment, spot calling, and cell segmentation diagnostics. I speak from hands-on runs where applying these metrics turned a flailing workflow into a predictable one: in one project (KL, Aug 2022) we reduced re-runs from 18% to 3% by changing the segmentation model and increasing local sequencing depth; that saved about two weeks of delay. I believe these metrics let you compare tools or tune Stereo-seq analysis workflow sensibly, not by hype. Final note — small pause — always validate with a known-control slide before committing to a full study.
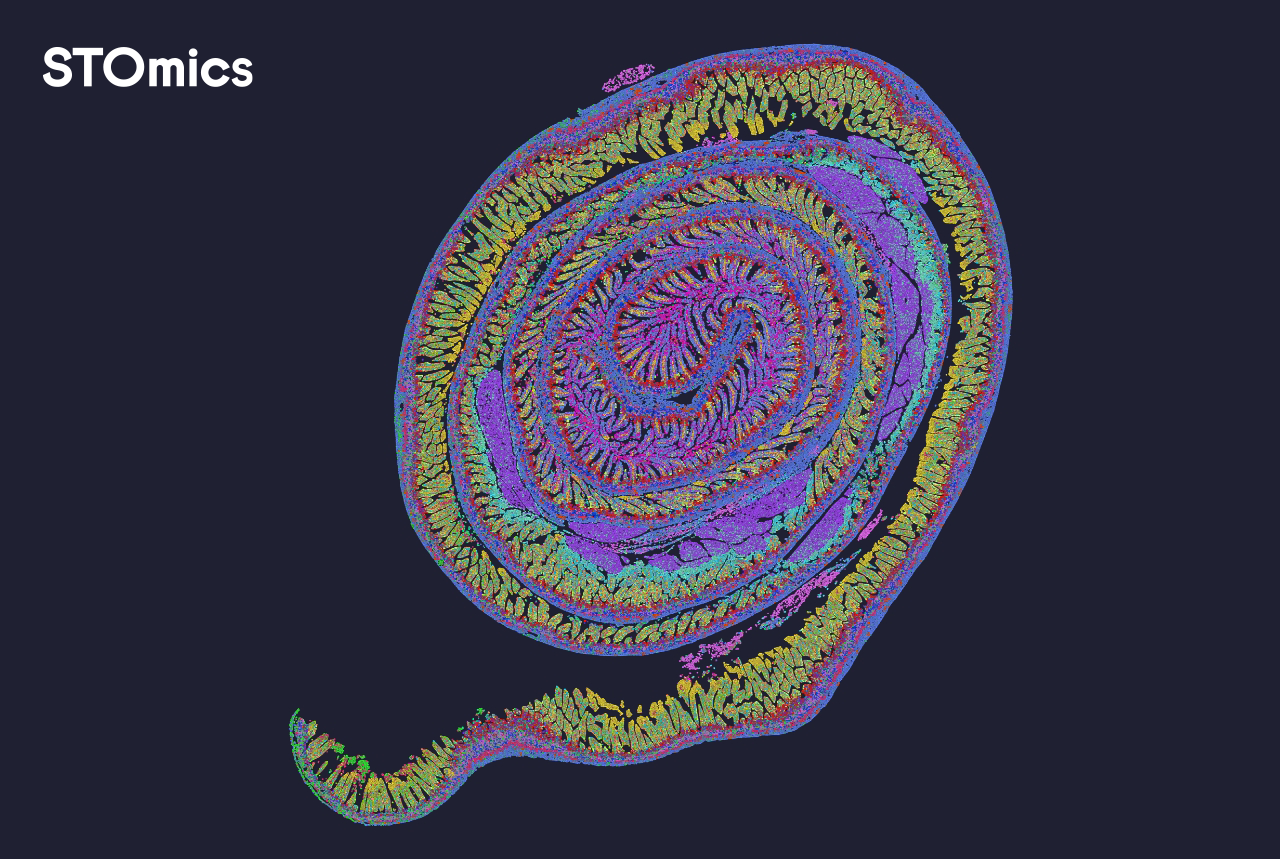
I’ve shared specific things I saw, the dates and sample types that taught me the hard way, and practical measures you can use; apply them, test, and you will cut wasted runs. For support and tools I still point colleagues toward stomics for practical integrations and clearer diagnostics: stomics.